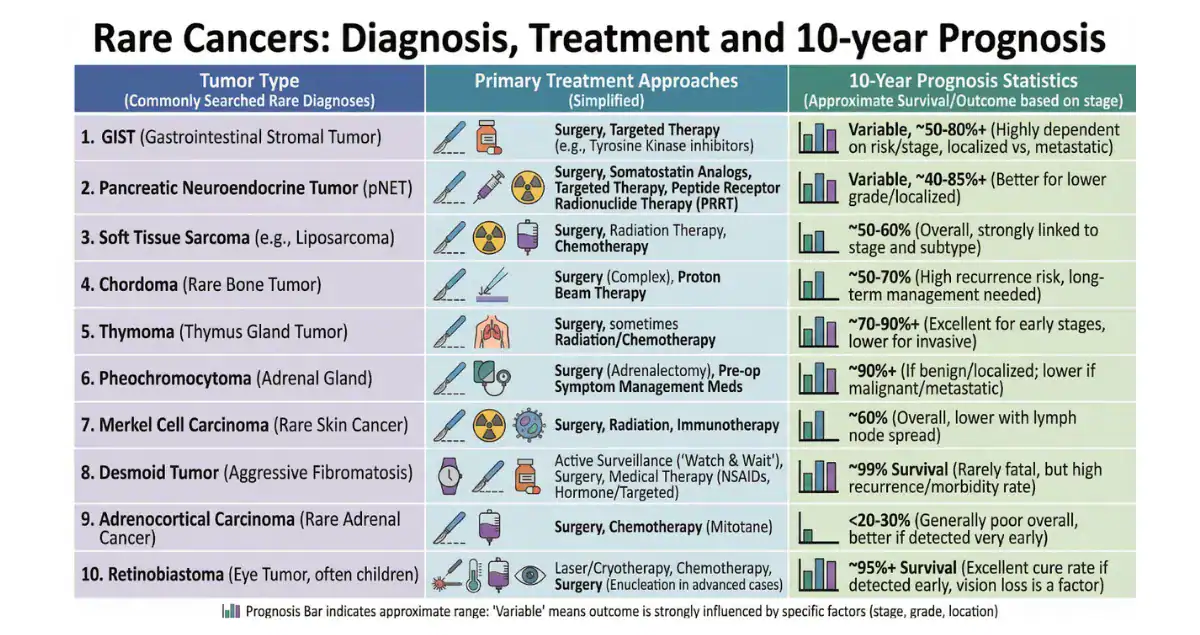
A cancer diagnosis is daunting enough, but when the tumor is rare or mimics everyday illnesses, the path to answers can stretch for months or years. Rare cancers,those affecting fewer than about 6 in 100,000 people annually,often evade early detection because their symptoms are vague, nonspecific, or resemble common conditions like infections, digestive issues, or musculoskeletal problems. Doctors may not encounter them frequently, leading to initial misdiagnoses or delayed referrals to specialists. Pathology becomes the turning point: a biopsy or surgical sample, examined with microscopy, immunohistochemistry, molecular testing, and sometimes genetic sequencing, finally uncovers the true identity. This guide focuses on a few particularly challenging rare cancers that patients and families frequently research after puzzling symptoms or unexpected pathology results. It explains why these tumors are tough to spot, what diagnostic clues pathologists look for, and how the process ultimately leads to clarity.
If any part of this explanation leaves you wanting more clarity, especially about your own pathology report, services like Honest Pathology offer virtual consultations with board-certified pathologists who review reports line by line, explain terms in plain language, and answer your specific questions directly.
Why Rare Cancers Are So Difficult to Diagnose
Rare cancers pose unique hurdles. Symptoms often appear late or mimic benign issues, so routine checkups rarely catch them. Imaging may show abnormalities that look like inflammation or cysts rather than malignancy. Because these tumors are uncommon, general pathologists may need second opinions or send samples to specialized centers for advanced testing. Molecular markers, unusual cell patterns, or specific protein expressions frequently provide the breakthrough. Delays mean the disease can advance before treatment starts, but accurate pathology diagnosis opens the door to targeted therapies, clinical trials, or specialized centers.
Sarcomas: The “Great Mimickers” of Soft Tissue and Bone Tumors
Sarcomas arise in bones, muscles, fat, cartilage, blood vessels, or connective tissues, over 70 subtypes exist, making them collectively rare (about 1% of adult cancers). They often present as painless lumps, swelling, or bone pain mistaken for sports injuries, arthritis, cysts, or lipomas. Early imaging might suggest benign growths, delaying biopsy. Pathology is essential: sarcomas show spindle-shaped or pleomorphic cells, specific staining patterns (e.g., for vimentin, desmin, or S100), and characteristic translocations like SYT-SSX in synovial sarcoma or EWS-FLI1 in Ewing sarcoma. Molecular testing identifies these fusions, distinguishing subtypes that respond differently to chemotherapy or targeted drugs. Diagnosis can take weeks due to the need for expert review, but once confirmed, referral to sarcoma centers improves outcomes.
Mesothelioma: The Asbestos-Linked Cancer That Hides in Plain Sight
Mesothelioma develops in the lining of the lungs (pleura), abdomen (peritoneum), or rarely heart/pericardium, strongly linked to past asbestos exposure with a latency of 20–50 years. Symptoms, shortness of breath, chest pain, abdominal swelling, or persistent cough, mimic pneumonia, heart failure, or gastrointestinal disorders. Fluid buildup on scans may be drained and tested, but malignant cells can resemble reactive mesothelial cells or adenocarcinoma. Pathology relies on biopsy showing epithelioid, sarcomatoid, or biphasic patterns, plus immunohistochemistry (positive for calretinin, WT-1, CK5/6; negative for TTF-1, CEA) to rule out lung cancer or metastases. Molecular markers like BAP1 loss help confirm. The process often requires multiple samples and expert consultation, explaining why diagnosis frequently occurs at advanced stages.
Neuroendocrine Tumors (NETs): The Slow-Growing “Silent” Cancers
NETs originate from hormone-producing neuroendocrine cells scattered throughout the body (most often gut, pancreas, or lungs). They are rare and notoriously slow-growing, with symptoms like flushing, diarrhea, abdominal pain, or wheezing dismissed as irritable bowel syndrome, food allergies, or anxiety. Many are incidental findings on scans for unrelated issues. Pathology is key: tumors show characteristic “salt-and-pepper” chromatin, positive staining for chromogranin A, synaptophysin, and often specific hormones. Ki-67 index grades proliferation rate (low-grade NETs vs. high-grade neuroendocrine carcinomas). Somatostatin receptor imaging (e.g., Ga-68 DOTATATE PET) aids detection, but tissue confirmation via biopsy is definitive. Delays of years are common before the right specialist connects vague symptoms to this diagnosis.
Adenoid Cystic Carcinoma: The Slow but Persistent Salivary Gland Cancer
This rare head and neck cancer (about 1% of such tumors) arises mainly in salivary glands but can occur in other sites like breast or trachea. It grows slowly with few early symptoms, often just a painless lump or facial numbness, mimicking benign salivary tumors or infections. Perineural invasion (spreading along nerves) causes pain or paralysis later. Pathology shows cribriform, tubular, or solid patterns with hyaline material and positive staining for c-KIT, p63, and MYB-NFIB fusion in many cases. The fusion is a hallmark detected by FISH or sequencing. Diagnosis may require multiple biopsies due to bland cytology on fine-needle aspiration, and expert head/neck pathologists are often consulted.
Appendix Cancer and Pseudomyxoma Peritonei: The Hidden Abdominal Threat
Primary appendix cancers, including mucinous neoplasms, are extremely rare and often discovered incidentally during appendectomy for presumed appendicitis. Symptoms like abdominal bloating, pain, or hernia mimic IBS or ovarian issues. When mucin-producing tumors rupture, they cause pseudomyxoma peritonei, a jelly-like buildup in the abdomen. Pathology examines the appendix for low-grade vs. high-grade mucinous neoplasms, checking for invasion, cellularity, and signet-ring cells. Immunohistochemistry (CK20+, CK7-) and molecular testing help classify. Diagnosis frequently occurs at surgery for “appendicitis,” with full staging requiring peritoneal exploration.
The Path to Diagnosis and Hope for Rare Cancers
Diagnosing these cancers often involves a circuitous route: initial misattribution to common ailments, inconclusive imaging, and repeated tests before biopsy confirms malignancy. Advanced pathology, immunohistochemistry, FISH, NGS, and expert review, provides the breakthrough, identifying actionable mutations or guiding specialized care. While rarity complicates early detection, it also drives dedicated research and centers of excellence. Many patients find answers through second opinions or rare-tumor programs.
If you or a loved one faces a puzzling pathology report or suspicion of a rare cancer, expert review can accelerate clarity. Consider consulting experts at Honest Pathology, who specialize in dissecting complex reports, whether unusual histology, rare markers, or molecular findings, and translating them into understandable, actionable insights for patients. This support empowers better discussions with specialists and a clearer path forward.